预测配体-靶标对的结合亲和力,哈工大开发新SOTA药物表示模型

编辑 | 白菜叶
了解配体-靶标对的分子间相互作用是指导优化癌症**研究的关键,这可以大大减轻湿实验室的负担。当前计算方法存在一些局限,限制了它们的实际应用。
在这里,哈尔滨工业大学的研究人员在此提出了 DrugMGR,这是一种深度多粒度**表示模型,能够够预测每个配体-靶标的结合亲和力和力和区域。
通过对配体复杂的自然机制和蛋白质**特征的多粒度表示学习,DrugMGR 几乎在所有数据集上都显示优于当前**进方法的效果。并且,这是**个同时使用图、卷积和基于注意力的信息分析蛋白质-配体复合物的模型。
该研究以「DrugMGR: a deep bioactive molecule binding method to identify compounds targeting proteins」为题,于 2024 年 4 月 1 日发布在《Bioinformatics》。

**开发对于疾病**至关重要,科学家们通过**再利用可以快速寻找**方案,但传统实验方法成本高、周期长,限制了其应用。相比之下,采用计算方法识别高可信度的配体-靶标相互作用,能够够显示著缩小选择范围,并揭示蛋白质-配体复合物的结合机制。
在过去十年中,生物活性分子的激增推动了深度学习和人工智能在研究蛋白质相互作用中的应用。这对于开发新**和理解疾病机制具有重要意义。
然而,现有深度学习方法存在两方面问题:一是多数模型对多粒度配体特征的捕获能力不足,未能充分整合原子环境、化学基因组序列等多元自然机制;二是许多方法忽视了对结合区域可解释性的构建,虽有少数借助注意力机制尝试推断结合位点,但关联生物特征不明确,不利于指导研究人员定位结合位点。
为了应对这些缺陷,哈尔滨工业大学的研究人员提出了 DrugMGR,这是一种基于深度多粒度表示的模型,可以预测配体与蛋白质靶标的结合亲和力和区域。
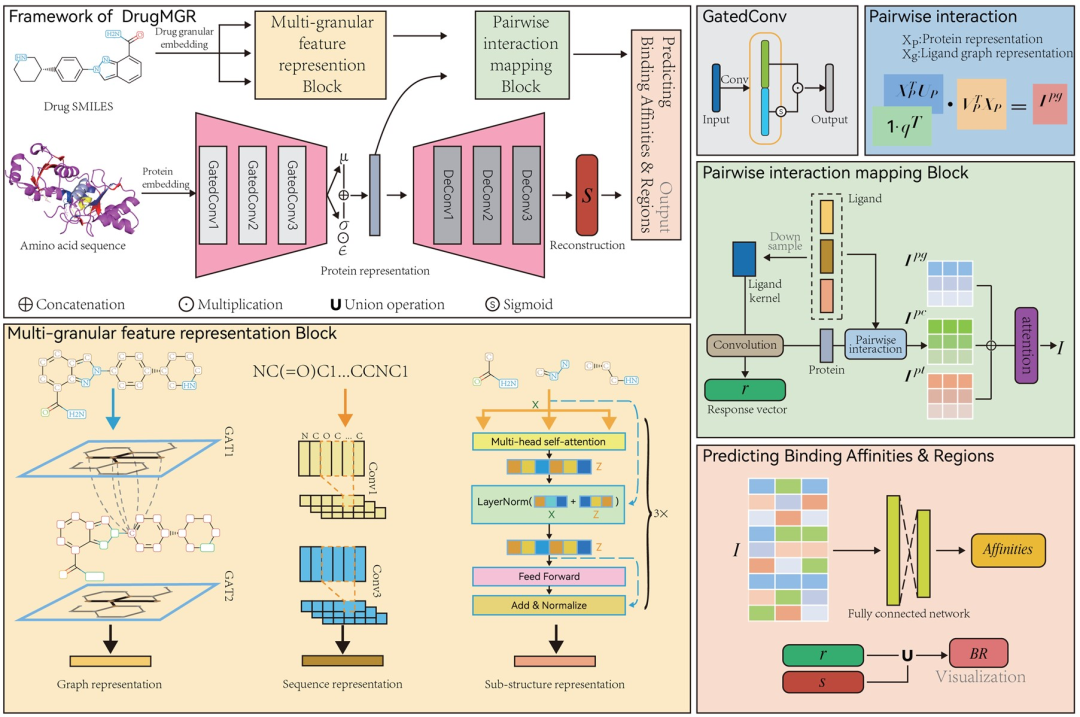
图示:DrugMGR 方法概述。(来源:论文)
具体来说,该团队首先使用三个深度模块来**编码配体的自然机制,即,用图注意力网络 (GAT) 来建模原子环境,用 CNN 来提取全局化学基因组序列,并使用分子Tran**ormer (MT) 来捕获局部子结构的相互影响。
研究人员还设计了一个并行 VAE 模块,通过 CNN 块在概率编码器中学习蛋白质的**特征,然后在概率解码器中重建目标结构。
然后,配体和蛋白质的编码表示被输入由注意力网络组成的成对相互作用映射模块,从而学习蛋白质-配体复合物的相互作用模式。联合成对相互作用表示由**连接的网络解码,用于预测生物活性分子的结合亲和力。
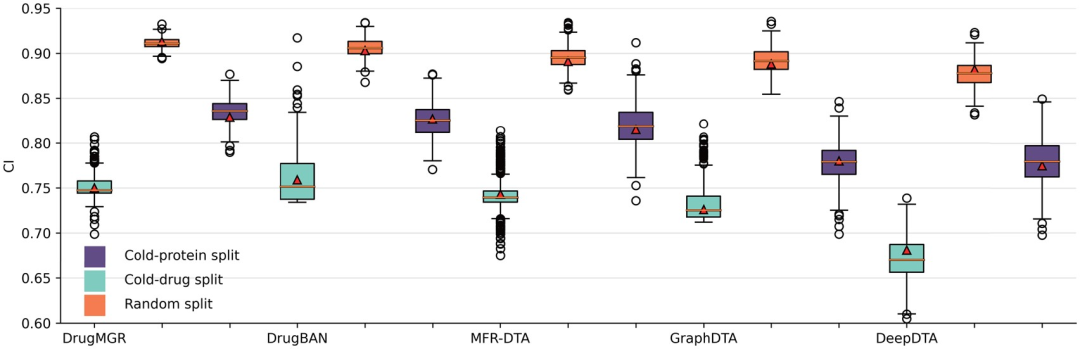
图示:BindingDB 数据集随机分割和冷启动分割的性能比较。(来源:论文)
对于结合区域预测,研究人员首先突出显示具有与配体结合潜力的重建蛋白质的结合位点,作为原始结合区域。随后,使用卷积运算将多粒度配体特征与蛋白质特征相乘。
接下来,他们将卷积结果记录为每个配体-靶标对的响应向量,并将响应向量中具有高值的区域标记为可视化结合区域。**,研究人员利用这两个区域来指导**预测的结合区域。
与 DrugBAN(一种用于简单识别**和靶标之间相互作用的二元分类器)相比,该团队提出的 DrugMGR 可以进一步了解蛋白质-配体复合物的综合结合信息(结合亲和力和结合区域),这在生物活性分子结合的实际应用中发挥着核心作用。

针对具有高度侵袭性、预后不良且缺乏有效靶向疗法的三阴性乳腺癌(TNBC),该团队利用 DrugMGR 模型从 DrugBank 数据库中识别针对 PARP1 的潜在抑制剂和化疗**。
筛选出的前 10 个候选化合物经 GeneCards 和 PDB 系统验证,并通过可视化 PARP1 与 Talazoparib(PDB ID: 4PJT)的结合区域,确认模型的有效性。
结果显示,DrugMGR 准确预测了结合位点,表现优异,有望成为针对 PARP1 虚拟筛选的**工具,助力生物医药学家筛选更优的抗肿瘤**组合。
论文链接:https://academic.oup.com/bioinformatics/article/40/4/btae176/7638803